
Dra. AM Caleya Zambrano / Dra. LT Altamirano Sánchez / Dra. C Cardoso Silva / Dra. M Maroto Edo / Dra. E Barbería Leache .Alumna del Máster de Odontopediatría. Colaboradora del Programa de Atención Odontológica a Pacientes en Edad Infantil. Departamento de Profilaxis, Odontopediatría y Ortodoncia. Facultad de Odontología. UCM./ Mster en Odontopediatría. Becaria del Programa de Atención Odontológica a Pacientes en Edad Infantil. Departamento de Profilaxis, Odontopediatría y Ortodoncia. Facultad de Odontología. UCM./ Profesora Asociada. Miembro del Equipo del Programa de Atención Odontológica a Pacientes en Edad Infantil. Departamento de Profilaxis, Odontopediatría y Ortodoncia. Facultad de Odontología. UCM. Catedrático de Odontopediatría. Directora del Programa de Atención Odontológica a Pacientes en Edad Infantil. Departamento de Profilaxis, Odontopediatría y Ortodoncia. Facultad de Odontología. UCM. Madrid.
Introducción
La osteogénesis imperfecta (OI) es una enfermedad grave cuyo rasgo principal es la fragilidad ósea. Esta característica hace que sea conocida popularmente como “la enfermedad de los huesos frágiles” (1-3). Esta patología se divide en varios tipos y, en algunos casos, incluye en su cuadro clínico la Dentinogénesis Imperfecta (DI).

Por tanto, el odontólogo clínico que diagnostique una dentinogénesis imperfecta tiene que determinar el tipo a que pertenece ya que la dentinogénesis imperfecta Tipo I se asocia a osteogénesis imperfecta. Si ésta no está diagnosticada, el odontólogo debe indicar a los padres la importancia de acudir al pediatra para que continúe las pruebas encaminadas a completar el diagnóstico de la enfermedad.
Dentinogénesis imperfecta
La Dentinogénesis Imperfecta es un trastorno en el desarrollo de la dentina de origen hereditario, de carácter autosómico dominante. Consiste en un defecto en la formación de la matriz orgánica de la dentina que se origina durante la fase de histodiferenciación (4-6). La dentina presenta un aspecto opalescente característico, por lo que también se ha denominado dentina opalescente hereditaria. La incidencia se estima alrededor de 1/8.000 (4-6). Shields dividió a la DI en tres grupos: la DI tipo I, la DI tipo II (o dientes de Capdepont) y la DI tipo III (Brandywine) (4, 8). La DI tipo II es la más común y la DI tipo III es la más rara (7, 9). La DI tipo I se asocia con la Osteogénesis Imperfecta, sin embargo los tipos II y III aparecen como entidades aisladas (4, 10).
La DI tipo I es una enfermedad genética, asociada a la osteogénesis imperfecta, causada por mutaciones en los genes que codifican el colágeno tipo I, causando una alteración en la formación de la dentina (11). Este tipo de Dentinogénesis puede orientar hacia un diagnóstico temprano de la OI mientras que los tipos II y III no tienen valor para este fin. Por tanto, se describirán a continuación las características de la DI tipo I y los datos necesarios, para realizar un diagnóstico diferencial con las del tipo II y III.
La DI tipo I es una enfermedad que aparece siempre asociada a OI. Sin embargo sólo entre un 10 a un 50% de los pacientes que padecen OI presentan DI (7, 12, 13). La relación se da con más frecuencia en los casos severos de OI (7, 8, 12-14).
La dentición temporal se afecta con más severidad que la permanente. En la dentición permanente las lesiones suelen ser menos graves e incluso, a veces, clínicamente indetectables. Las afectaciones son mayores en los dientes formados en estadios tempranos. (14, 15).
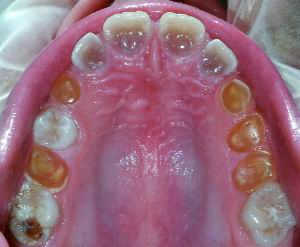 |
| Figura 1. Imagen clínica de Dentinogénesis Imperfecta, en la arcada superior, donde se puede observar un gran desgaste de los dientes temporales y la alteración de color de todos ellos |
Manifestaciones clínicas de la Dentinogénesis
Imperfecta tipo I
En la Dentinogénesis de este tipo, las coronas de los dientes de ambas denticiones presentan un color ámbar translúcido y opalescente (Figura 1) (8, 9, 13). Existen alteraciones del color, presentando un color variable amarillo, azulado, marrón o gris (2, 4, 5, 7, 12, 14-18). La morfología coronaria ha sido descrita de múltiples formas: grandes, cortas, normales, abrasionadas, con forma de campana, o presentando varias de estas características (7-19).
El esmalte sobre la dentina afectada tiende a “astillarse” por el borde incisal de dientes anteriores, por el surco oclusal de dientes posteriores y por los surcos linguales y bucales de todos los dientes (15), debido a ello la dentina queda expuesta y sufre un desgaste muy rápido que puede llegar a ser muy grave (2, 7, 15). Por este motivo se ha denominado a esta enfermedad “dientes sin coronas” (4, 8, 20) y como resultado se puede producir una reducción de la dimensión vertical (Figura 2 a, b) (21).
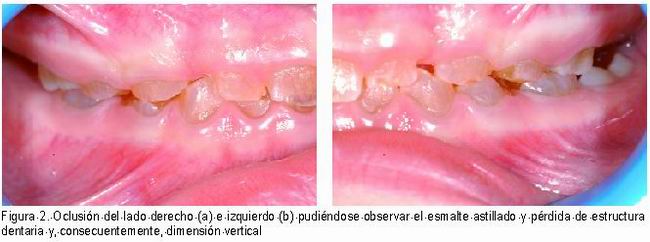 |
Características radiográficas de la Dentinogénesis Imperfecta tipo I
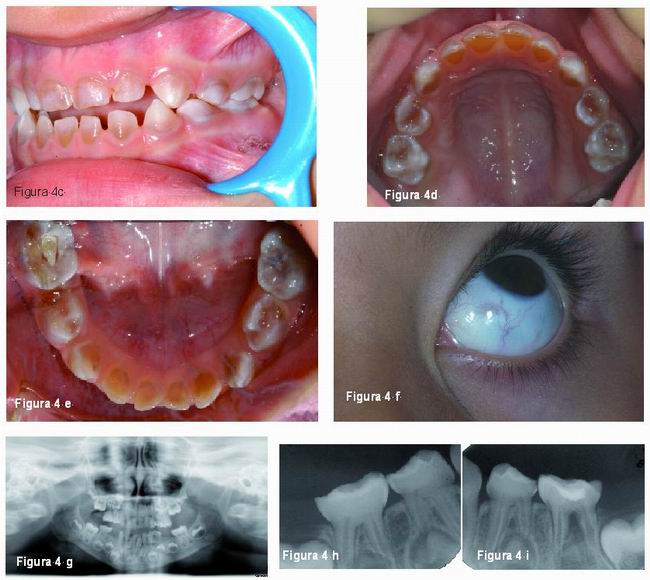
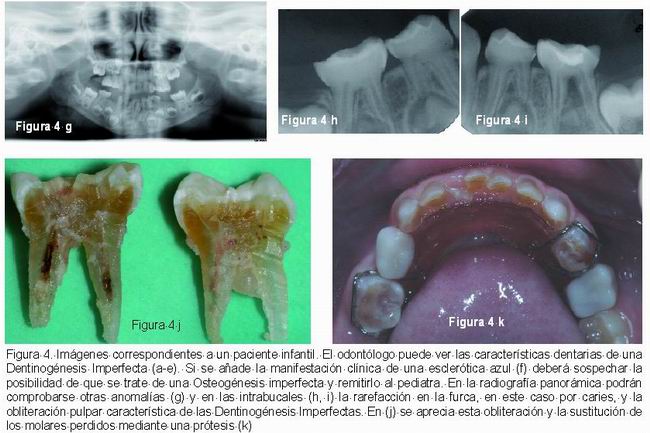
Radiográficamente, las coronas dentarias son bulbosas, con una constricción muy marcada a nivel cervical, raíces cortas y delgadas (16), reducción del tamaño u obliteración de la cámara pulpar que puede afectar también al sistema de conductos radiculares (4, 6, 7, 14, 15).Esta obliteración pulpar ocurre tanto en los dientes temporales como en los permanentes jóvenes, los cuales sufren una obliteración pulpar acelerada. Algunos autores refieren que esta obliteración puede ocurrir justo cuando erupcionan e incluso antes de erupcionar, aunque lo frecuente es que ocurra después de la erupción(Figura 3) (8, 13-16).
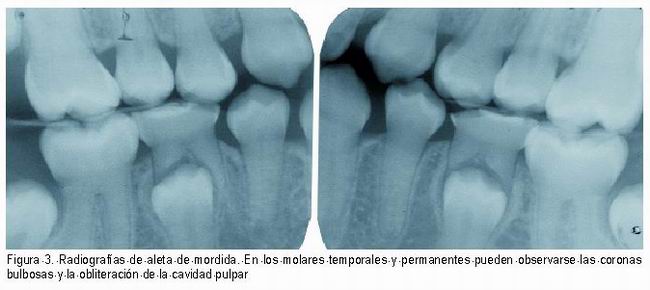 |
Schwartz and Tsipouras comprobaron que la rapidez de la obliteración pulpar puede ser diferente dependiendo del tipo de OI que se padezca, siendo más rápida en la OI tipo III (14).
McDonald (5) refiere que ocasionalmente pueden observarse rarefacciones apicales en dientes temporales aunque no propone ninguna explicación satisfactoria para esta alteración. También otros autores como Huber y Tanaka refieren hallazgos similares (19, 22).
Histopatología de la Dentinogénesis Imperfecta tipo I
El hallazgo histológico más frecuente es la presencia de dentina anormal (7, 15), pudiendo existir zonas atubulares, y zonas con túbulos en número reducido y tamaño variable (2, 4, 7, 8, 13, 15, 23, 24). Lindau et al (24) observaron que la mayoría de las alteraciones de la dentina se producen cerca del esmalte por lo que sugieren que estas anomalías dentinarias se originan en estadios tempranos de la Dentinogénesis (7). Además de las alteraciones anteriores en numerosos estudios histológicos se observó que el colágeno tipo I en la dentina circumpulpar es generalmente escaso y las fibras de colágeno pueden estar incompletamente mineralizadas (25).
Las características del esmalte y de la unión amelodentinaria son controvertidas ya que algunos autores defienden que no se hallan anormalidades en el esmalte de los pacientes afectados (7, 15). Sin embargo, otros investigadores han encontrado alteraciones principalmente en la unión amelodentinaria, debidas a que los daños que se producen en el mesodermo en la OI, también podrían afectar al tejido ectodérmico del esmalte durante la amelogénesis (2). Estos hallazgos histológicos podrían explicar que en los mecanismos de fractura del diente estarían implicados no sólo los defectos dentinarios sino también los de la unión amelodentinaria (2, 24, 26).
En el interior de la cámara pulpar es frecuente encontrar cuerpos calcificados o dentículos verdaderos (4, 8), los cuales poseen túbulos dentinarios (27), pero es discutido si su presencia es normal o patológica (2, 8, 13, 24).
Diagnóstico diferencial entre los diferentes tipos
La Dentinogénesis Imperfecta tipo I y tipo II tienen hallazgos clínicos, radiológicos e histológicos comunes (8, 17, 19), sin embargo pueden describirse hallazgos diferenciadores entre ambos tipos. Entre estos, los más relevantes son:
— En el árbol genealógico de los niños con Dentinogénesis tipo II no se encuentran familiares con Osteogénesis, sin embargo pueden encontrarse en la tipo I.
— La afección dentaria en la tipo II tiene el mismo grado de severidad en todos los miembros de la familia y en la DI tipo I muestran diferencias.
— En la tipo II ambas denticiones están afectadas, mientras que en la DI tipo I se afectan más los dientes temporales que los permanentes (8, 9, 16).
— La DI tipo III es extremadamente rara (10) estando limitada su presentación al área geográfica de Maryland. No suele presentar rarefacciones periapicales (8, 10).
Osteogénesis imperfecta
La osteogénesis imperfecta (OI) es un trastorno hereditario autosómico dominante que afecta principalmente a las estructuras formadas por colágeno tipo I. Es conocida con múltiples denominaciones en el ámbito médico, entre ellas: fragilidad ósea, Síndrome de Lobstein o Síndrome de Van Der Hoeve, y a medida que aumentan las investigaciones se van proponiendo nuevas denominaciones y clasificaciones (28).
La frecuencia de presentación de este desorden oscila entre 1:5.000 a 1:20.000 nacidos, según los diferentes autores (1, 19, 29, 30).
Etiología
La mayoría de los tipos de OI están asociados con una mutación en los genes COL1A1 y COL1A2 que se encuentran en los cromosomas 17 y 7 respectivamente, los cuales codifican al colágeno tipo I (7, 17, 20, 30). Esta mutación produce alteraciones cualitativas o cuantitativas del colágeno tipo I, afectando de forma directa a diferentes estructuras como hueso, articulaciones, ojos, piel y dientes (1, 14, 16, 28, 31). Se considera que en las OI leves (tipo I) lo que se produce es un defecto cuantitativo del colágeno tipo I, mientras que en las OI moderadas y severas se produce un defecto cualitativo o estructural .Cuanto más importante es la afectación del colágeno, mayor es el grado de afectación de las estructuras que lo contienen (7, 13).
Clasificación
Clásicamente se diferenciaban dos formas clínicas de OI: “Congénita”, la cual podía ser diagnosticada radiográficamente en el útero por la presencia de múltiples fracturas y producía la muerte intraútero o poco después del nacimiento, o “Tardía”, que se detectaba en la infancia y solía ser más leve (1, 7, 18, 28).
Sillence et al. diferenciaron la OI en cuatro grupos basándose en criterios clínicos, hereditarios y radiográficos. Ellos identificaron la OI tipo I (o Leve) de carácter autosómico dominante; la OI tipo II (o Perinatal Mortal) como autosómica recesiva; la OI tipo III (o Deformante progresiva) engloba un grupo de casos esporádicos probablemente heterogéneo desde el punto de vista clínico y genético; la OI tipo IV (o Moderadamente grave) como enfermedad autosómica dominante (1, 7, 12).
Recientemente, nuevos avances histológicos y genéticos han permitido la clasificación de varios pacientes con OI tipo IV en 3 nuevos tipos: tipo V (Callo Hiperplásico), tipo VI y tipo VII (7, 19).
Además, la OI de los tipos I y IV se subdividen en subtipos A y B en función de la ausencia o presencia de dentinogénesis imperfecta. Y se ha propuesto recientemente que esta subdivisión también es válida para los tipos II y III ya que en éstos también se ha observado que aparece Dentinogénesis Imperfecta (12, 14, 16).
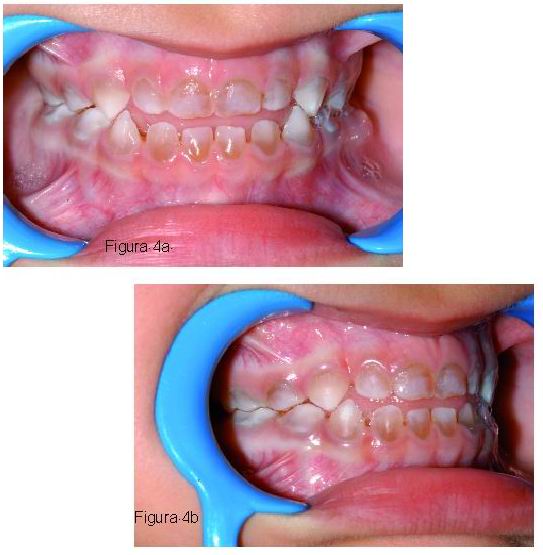 |
Manifestaciones clínicas
Las manifestaciones clínicas generales de la OI se caracterizan por una fragilidad ósea, osteoporosis, múltiples fracturas de huesos largos, estatura baja y progresiva deformidad esqueléticas. Otras manifestaciones que pueden estar presentes son: escleróticas azules, sordera temprana, hiperlaxitud de las articulaciones y ligamentos, piel delgada y laxa y dentinogénesis imperfecta (1-3, 7, 12, 28, 29). Es frecuente encontrar la triada de sordera, esclerótica azul y fragilidad ósea (Figura 4) (1, 28).
Sin embargo cada tipo de OI presenta rasgos clínicos diferenciadores:
En la OI tipo I (Leve) se manifiesta fragilidad ósea leve, escleróticas azules, pérdida de audición y estatura baja (7, 12, 15).
En la OI tipo II (Perinatal Mortal) hay fragilidad ósea extrema, fracturas intrauterinas, y suele producir la muerte intrauterina o perinatal (7, 12).
En la tipo III (Deformante Progresiva) la fragilidad ósea es severa pero compatible con la vida. Es progresivamente deformante, hay severa osteoporosis, macrocefalia con fascies triangulares, escoliosis, las escleróticas son ligeramente azules y la DI es común (7, 12). En este tipo, además es más frecuente encontrar estatura muy baja (15, 21).
Tipo IV (Moderadamente Grave): diferentes niveles de fragilidad ósea, y la esclerótica es normal o ligeramente azul (7, 12).
Tipos V (Callo Hiperplásico), VI y VII: el hueso presenta distintos características clínicas, radiológicas e histológicas, pero no presentan escleróticas azules ni DI (7, 12).
Hallazgos orales y craneofaciales
Las características craneofaciales que pueden ser observadas en pacientes con OI son una base craneal aplanada, morfología craneofacial triangular, una frente especialmente ancha y abombada, proyección de los pabellones auriculares, disminución de la dimensión vertical y occipucio sobresaliente (3, 21, 28).
En cuanto a los hallazgos orales, la manifestación más característica de la OI es la dentinogénesis imperfecta tipo I (2, 19). Cuanto más severa sea la OI, es más probable que ésta se asocie con DI (12). Además han sido halladas otras alteraciones orales asociadas a la OI como maloclusión, agenesias (17, 28), anomalías dentarias caracterizadas por extensión apical de las cámaras pulpares que recuerdan al taurodontismo (17), impactación del primer y/o segundo molar, y a veces de los caninos superiores (3, 14, 17, 31), erupción ectópica del primer y segunda molar permanente. (15). En algunos casos se han observado variaciones en la maduración dentaria en OI tipo III y OI tipo IV respectivamente (15). Esto demuestra que el hecho de que un paciente con OI no presente DI, no indica la ausencia de otras alteraciones dentales (13).
En cuanto a las alteraciones de la oclusión, se ha detectado oclusión de Clase III en pacientes con OI (3, 14, 15, 31), observándose en un 66, 6% de los pacientes estudiados (14). Además algunos autores describen un alto porcentaje de mordidas cruzadas posteriores unilaterales o bilaterales(14, 31). En general, cuanto más severa es la OI mayor es la posibilidad de maloclusión (14).
 |
 |
Manejo clínico de la Dentinogénesis Imperfecta y consideraciones en atención dental de pacientes con Osteogénesis Imperfecta
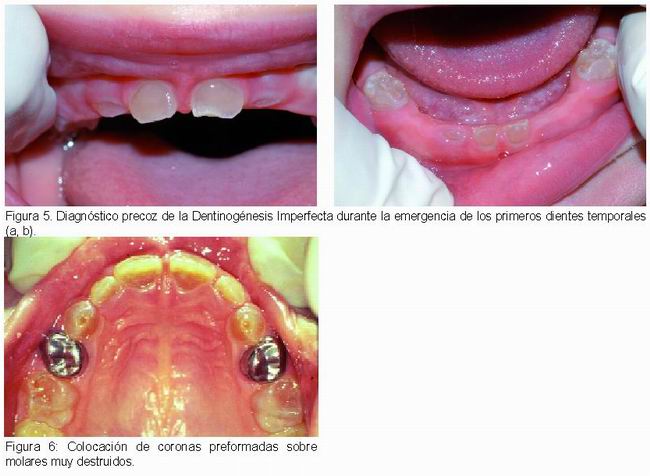
El tratamiento de la dentinogénesis imperfecta es muy complejo. Dado que las manifestaciones ocurren desde la emergencia de los primeros dientes temporales (Figura 5 a, b), a la complejidad del tratamiento, tanto en dentición temporal como en permanente, se añade la dificultad del manejo del niño y las implicaciones sobre el desarrollo de la autoestima y el proceso de socialización (5).
Han sido propuestas diferentes alternativas de tratamiento, todas ellas con resultados muy limitados. En el pasado, los dientes se solían extraer (7). Actualmente las propuestas de tratamiento consisten en procedimientos preventivos para evitar el desgaste (32), colocación de selladores, aplicación de flúor e instrucciones de higiene oral (7, 33). Los procedimientos restauradores en dentición temporal requieren plantearse la utilización de coronas de acero inoxidable (Figura 6) si la morfología de los molares afectados lo permite. Frecuentemente esto no es posible y hay que utilizar sobredentaduras para mantener la dimensión vertical y mejorar la estética (7, 5, 33).
 |
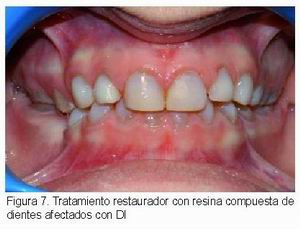 |
En dentición permanente joven se emplean resinas compuestas (Figura 7) y cemento de vidrio ionómero, y en pacientes adultos se suelen utilizar, además, carillas y coronas de recubrimiento total. Aunque en teoría la unión del adhesivo a la estructura dentaria es comprometida y puede producir fracasos de las restauraciones, también se ha comprobado que puede ser un éxito clínico en muchos pacientes y por ello se sigue utilizando en pacientes con DI (7, 15). En dientes con grandes destrucciones, rarefacción apical y fracturas radiculares puede ser necesaria la extracción (7, 15).
Por otro lado, si la historia clínica confirma la presencia de osteogénesis imperfecta, habrá que tener especial cuidado durante la realización de los tratamientos. La facilidad de fractura de los huesos obliga a ser cuidadosos con la postura del paciente en el sillón dental, debiendo ser segura y confortable (15). Durante las exodoncias debemos tener un extremo cuidado con el riesgo de producir fracturas mandibulares y fracturas dentarias, estas últimas debidas a la presión del fórceps (28, 32). Además, la alteración del tejido conectivo puede producir, en algunos pacientes, vasculopatías y trastornos de la hemostasia y de la cicatrización, con el consiguiente riesgo de hemorragias (3, 31, 32). La administración de profilaxis antibiótica para la realización del tratamiento dental estará determinada por la historia médica del paciente (15, 19). Por último se ha observado una alta incidencia de alergia al látex en algunos pacientes debido al elevado número de intervenciones médicas y quirúrgicas a las que han podido ser sometidos (15, 19).
Conclusiones
En la Osteogénesis Imperfecta es posible que las alteraciones dentales sean las primeras en ser detectadas como un cuadro de Dentinogénesis Imperfecta, por tanto el odontólogo debe estar capacitado para saber detectarlas y derivar al paciente al especialista para permitir un diagnóstico temprano de la enfermedad.
En los casos de Dentinogénesis Imperfecta, será muy importante realizar un correcto diagnóstico diferencial con otras anomalías dentarias y así poder instaurar un adecuado programa preventivo y restaurador.
Correspondencia
E. Barbería Leache
Dpto. Estomatología IV. Facultad de Odontología.
Plaza de Ramón y Cajal, s/n. 28040 Madrid
Bibliografía
1. Behrman, Kliegman, Jenson. Nelson. Tratado de pediatría. 17ª ed. Barcelona: Elseiver; 2004.
2. Lindau BM, Dietz W, Hoyer I, Lundgren T, Storhaug K, Norén JG. Morphology of dental enamel and dentine-anamel junction in osteogénesis imperfecta. Int J Paediatr Dent.1999; 9:13-21.
3. Kindelan J, Tobin M, Roberts-Harry D, Loulota RA. Orthodontic and orthognathic management of a patient with osteogenesis imperfecta and dentinogenesis imperfecta: a case report. J Orthod. 2003; 30.291-6.
4. García Barbero J. Patología y terapéutica dental. Madrid: Síntesis; 1997.
5. McDonald RE, Avery DR. Odontología pediátrica y del adolescente. 6ª ed. Madrid: Mosby/Doyma Libros;1995.
6. Cauwels RGEC, De Coster PJ, Mortier GR, Marks LAM, Martens. Dentinogenesis imperfecta associated with short stature, hearing loss and mental retardation: a new syndrome with autosomal recessive inheritance? J Oral Pathol Med. 2005; 34: 444-6.
7. Rios D, Vieira AL, Tenuta LM, Machado MA. Osteogénesis imperfecta and Dentinogénesis imperfecta: associated disorders. Quintessence Int. 2005; 36 (9):695-701.
8. Shields ED, Bixler D, El-Kafrawy. A proposed classification for heritable human dentine defects with a description of a new entity. Archs Oral Biol. 1973; 18:543-53.
9. Kamboj M, Chandra A. Dentinogenesis imperfecta type II: an affected family saga. J Oral Sci. 2007; 49(3): 241-244.
10-Barbería Leache E, Boj Quesada JR, Catalá Pizarro M, Garcia Ballesta C, Mendoza Mendoza A. Odontopediatría. 2ª ed. Barcelona: Masson; 2001.
11. Malmgren B, Lindskog S. Assessment of dysplastic dentin in osteogenesis imperfecta and dentinogenesis imperfecta. Acat Odontol Scand. 2003; 61(2):72-80.
12. Petersen K, Wetzel WE. Recent findings in classfication of osteogenesis imperfecta by means of existing dental symptoms. J Dent Child. 1998; 65(5):305-9.
13. Lund AM, Jensen BL, Nielsen LA, Skovby F. Dental manifestation of osteogenesis imperfecta and abnormalities of collagen I metabolism. J Craniofac Genet Dev Biol. 1998; 18(1):30-7.
14. Schwartz S, Tsipouras P. Oral findings in osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol. 1984; 57(2):161-7.
15. O´Connell AC, Marini JC. Evaluation of oral problems in an osteogenesis imperfecta population. Oral Surg Oral Med Oral Pathol Radiol Endod. 1999; 87: 189-96.
16-Ferraris ME, Carranza M, Fili T, Ferraris R. Defectos estructurales de los tejidos dentarios en desarrollo asociados a la Osteogénesis imperfecta letal (tipo II). Av Odontoestomatol. 1998; 14(6):383-90.
17-Malmgren B, Norgren S. Dental aberrations in children and adolescents with osteogenesis imperfecta. Acta Odontol Scand. 2002; 60(2):65-71.
18. Levin LS, Salinas CF, Jorgenson RJ. Classification of osteogenesis imperfecta by dental characteristics. Lancet. 1978; 1 (8059):332-3.
19. Huber MA. Osteogenesis imperfecta. Oral Surg Oral Med Oral Pathol Radiol Endod. 2007; 103(3):314-320.
20. Malmgren B, Lindsokg. Assesmente of dysplastic dentin in osteogénesis imperfecta and dentinogenesis imperfecta. Acta Odontol Scand. 2003; 61(2): 72-80.
21. Jensen BL, Lund AM. Osteogenesis imperfecta: Clinical, cephalometric, and biochemical investigations of OI types I, III, and IV. J Craniofac Genet Dev Biol. 1997; 17: 121-32.
22. Tanaka T, Murakami. Radiological features of hereditary opalescent dentin. Dentomaxillofac Radiol. 1998 ; 27:251-3.
23. Sunderland EP, Smith CJ. The teeth in osteogenesis and dentinogenesis imperfecta. Br Dent J. 1980; 149(10):287-9.
24. Lindau BM, Dietz W, Hoyer I, Storhaug K, Norén JG. Discrimination of morphological findings in dentine from osteogenesis imperfecta patients using combinations of polarized light microscopy, microradiography and scanning electron miscroscopy. Int J Paediatr Dent.1999; 9:253-261.
25. Waltimo J. Hyperfibers and vesicles in dentin matrix in dentinogenesis imperfecta (DI) associated with osteogenesis imperfecta (OI). J Oral Pathol Med. 1994; 23(9):389-93.
26. Hu JC-C, Simmer JP. Developmental biology and genetics of dental malformations. Orthod Craniofacial Res. 2007; 10:45-52.
27. Gómez de Ferraris ME, Campos Muñoz A. Histología y embriología bucodental. 2ª ed. Madrid: Editorial Médica Panamericana;2002
28. Manrique Mora C, González Márquez I, Peñalver Sánchez MA. Osteogenesis imperfecta: presentation of a case. Av Odontoestomatol. 1991; 7 (3):189-92.
29. Engelbert RH, Uiterwaal CS, Gulmans VA, Pruijs H, Helders PJ. Osteogenesis imperfecta in childhood: prognosis for walking. J Pediatr. 2000; 137(3):397-402.
30. Antoniazzi F, Mottes M, Fraschini P, Brunelli PC, Tató L. Osteogenesis Imperfecta. Practical Treatment Guidelines. Paediatr Drugs. 2000; 2(6)65-88.
31. Bullón P, Machuca G. Tratamiento odontológico en pacientes especiales. Madrid; 2004.
32. Prabhu N, Duckmanton N, Stevenson A, Cameron A. The placement of osseointegrated dental implants in a patient with type IV B osteogenesis imperfecta: a 9-year follow-up. Oral Surg Oral Med Oral Pathol Oral Radiol Endod.2007; 103:349-54.
33. De Nova Garcia MJ, Planells del Pozo P, Costa Ferrer F, Barberia Leache E. El odontopediatra ante la Dentinogénesis imperfecta. Av Odontoestomatol. 1991; 7(3):173-177.
e-mail: barberia@odon.ucm.es


